Omics concepts
This set of concepts is created to represent different types of omics data. The concepts cover clinical use cases from oncology, paediatric care, and pathogen surveillance.
Note
To find out more about the choices behind the modeling and concept design, have a look at the following publication:
van der Horst, E.; Unni, D.; Kopmels, F.; Armida, J.; Touré, V.; Franke, W.; Crameri, K.; Cirillo, E.; Österle, S. Bridging Clinical and Genomic Knowledge: An Extension of the SPHN RDF Schema for Seamless Integration and FAIRification of Omics Data. Preprints 2023, 2023120373. https://doi.org/10.20944/preprints202312.0373.v1
Concept design
The concepts allow to describe metadata on the omics process, and, in combination with other
concepts, the outcome of the genomics workflow. Also bulk transcriptomics and, to a lesser
extent, omics research in general can be represented. Each step in the omics workflow is a process
concept that is composed of essential metadata about that process. The three top-level concepts for
representing the omics workflow are Sample Processing, Assay, and Data Processing. The
Sample Processing and Data Processing concepts are processes that are executed on some input to
generate some output. The Sample Processing concept can have zero or more input Sample and zero
or more output Sample. Similarly, the Data Processing concept can have zero or more input
Data File and zero or more output Data File.The Sample Processing concept is composed of
zero or more input and output Sample concepts, while the Data Processing concept is composed
of zero or more input and output Data File concepts. The Assay concept, also a process, can
have zero or more input Sample and zero or more output Data File which indicates that the
Assay is a process that transforms an input Sample to an output Data File. As part of the
concept design, we also provided a way to express multi-step processes via the hasPredecessor
property. For example, a process B can link to process A that occurred before itself via the
hasPredecessor property. This style of representation is applicable for Sample Processing,
Data Processing, and in some cases, the Assay concepts.
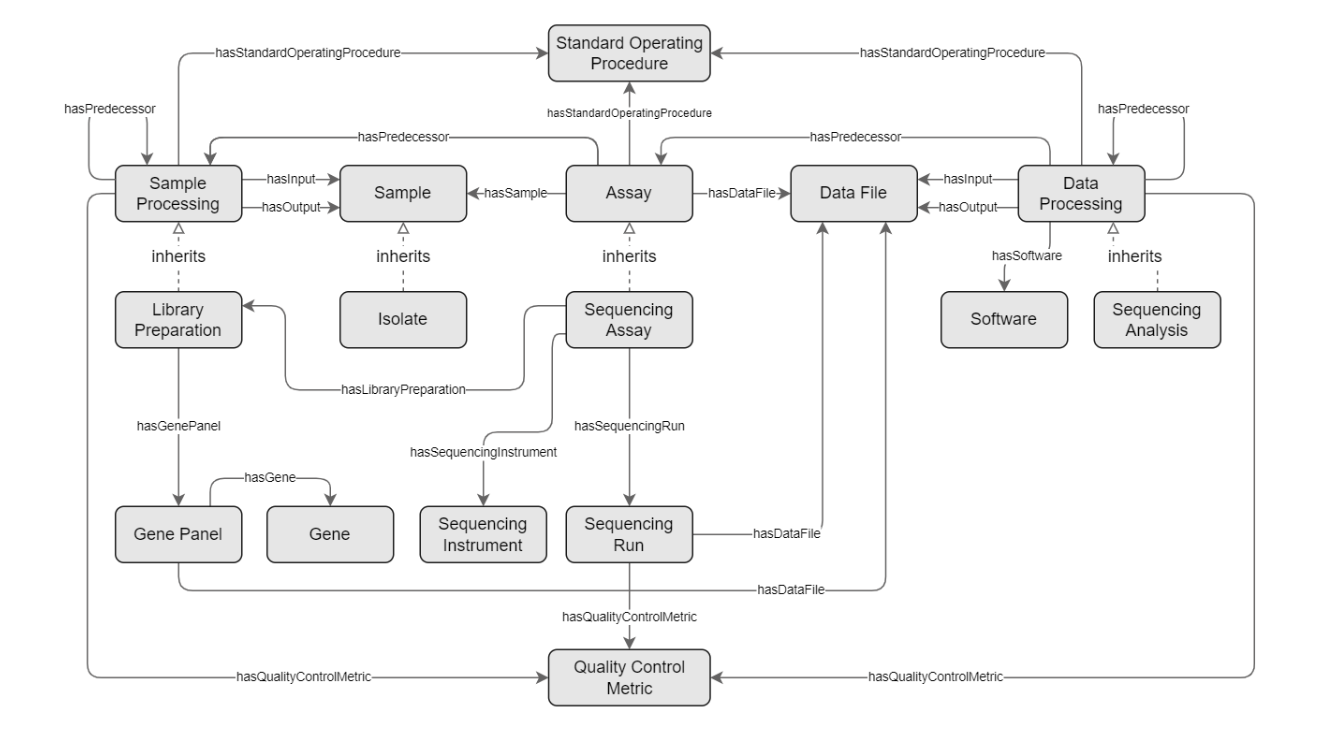
Figure 1. An overview of the (gen)omics concepts
Examples of data delivery
In this section each of the concepts are explained in more detail and examples will be provided.
Assay
Assay metadata is essential when sharing (experimental) results: for providing context, ensuring data quality, enabling data integration, and facilitating collaboration and reproducibility in research and clinical settings. An Assay takes a sample and produces data about that sample. For different types of omics research, different types of assays will be relevant, each with their own defining attributes. The Assay concept can be used as-is, or inherited by more specific types of assay.
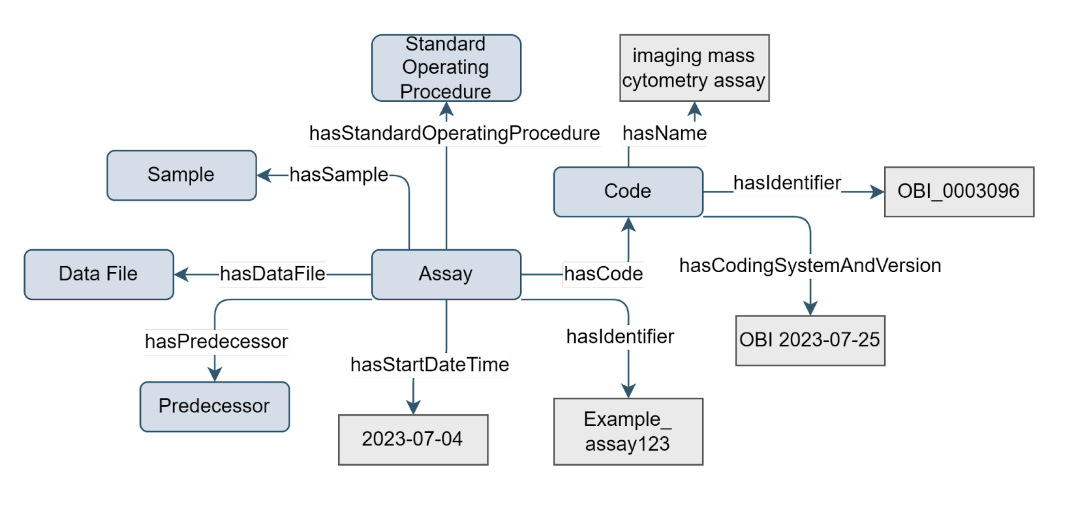
Figure 2. Example of the Assay concept
Guidelines for data delivery
The Assay concept has a
hasCodeproperty which can have a value that is a descendant ofOBI:0000070 | assay |or otherWhen multiple runs are executed for the same assay, as is the case for Whole Genome Sequencing, the start datetime for this concept will be equal to the run datetime of the Run concept that was first executed
All the properties are optional, except for the
hasCodeproperty
Sequencing Assay
Central to the genomics workflow is the sequencing assay. The Sequencing Assay concept is composed of essential metadata, representing the sequencer (via Sequencing Instrument), library preparation (via Library Preparation), intended read length and depth, and zero or more runs (via Sequencing Run). The Sequencing Assay concept is a type of Assay.
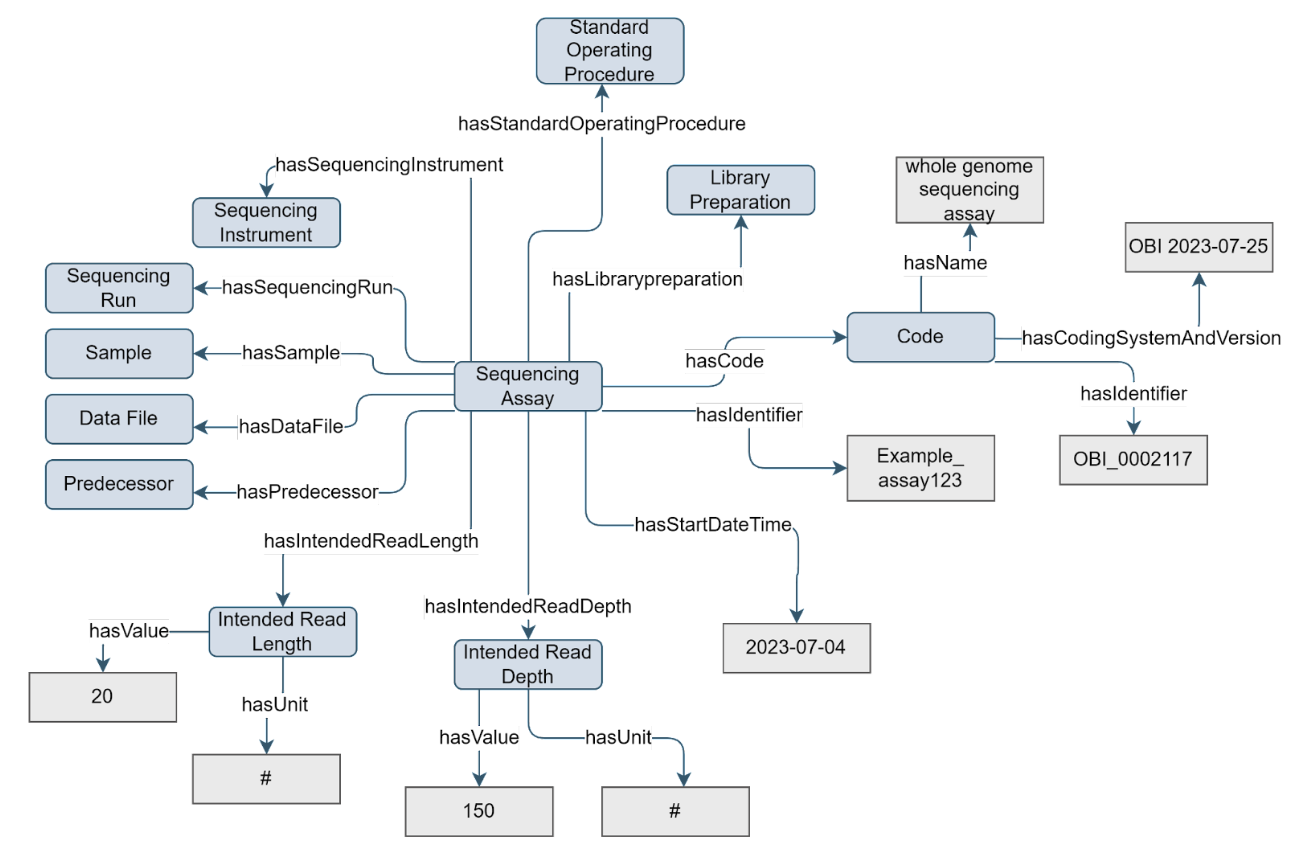
Figure 3. Example of the Sequencing Assay concept
Guidelines for data delivery
A Sequencing Assay may produce multiple Data Files, either different files from a single run or from multiple runs. It is possible to define run-specific information using Sequencing Run, or leave this information out. If a Data File is produced by a Sequencing Run, it follows that it is also related to the linked Sequencing Assay
When multiple runs are executed for the same Sequencing Assay, the start datetime for this concept will be equal to the run datetime of the run that was first executed The Sequencing Assay concept has a hasCode property which can have a value that is a descendant of
OBI:0000070 | assay |or other
Sequencing Run
The Sequencing Run concept represents the actual execution of the assay, and holds information that may vary per run, such as read count, average insert size, average read length, and quality control metrics (represented via the Quality Control Metric concept).
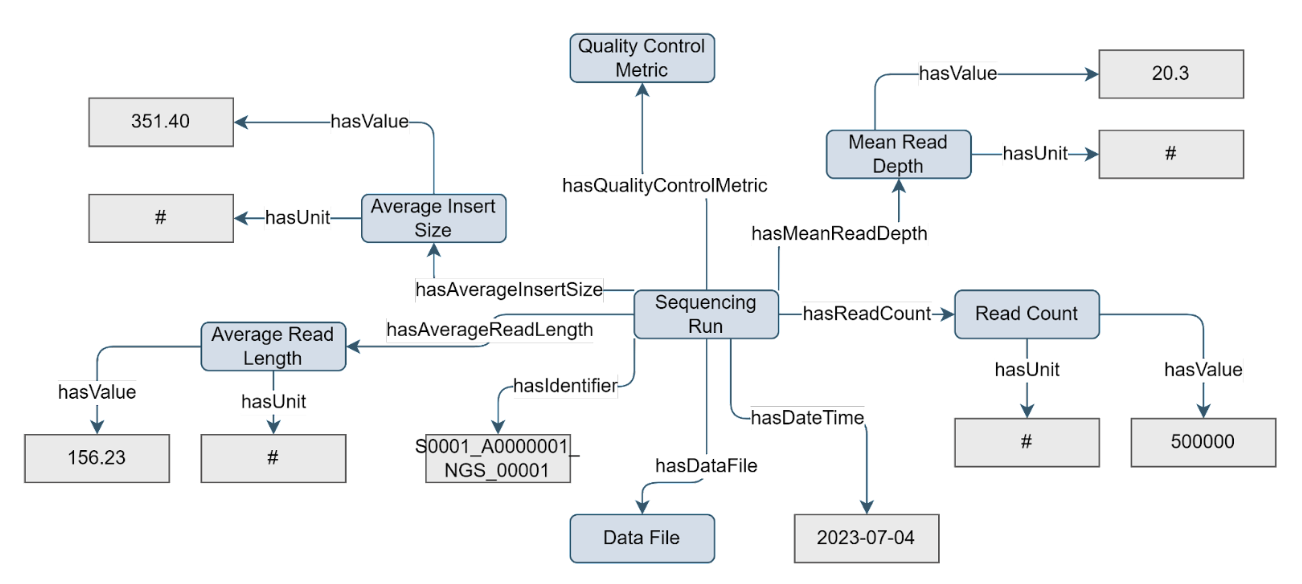
Figure 4. Example of the Sequencing Run concept
Guidelines for data delivery
At least one Data File and Quality Control Metric must be specified. As a result the cardinality for
hasDataFileandhasQualityControlMetricis1:n
Sequencing Instrument
The instrument that was used to conduct a sequencing assay is essential information to understand and evaluate the experimental context and data generation process. Sequencing data may be generated using a range of instruments. Different instruments may vary in sensitivity, accuracy, or precision, and recording the instrument used allows researchers to assess data quality and identify potential sources of variability. Knowing the instrument that was used to produce a dataset enables researchers to assess compatibility and potential biases when performing cross-platform comparisons. The Sequencing Instrument concept contains information about the instrument that was used to conduct a sequencing assay.
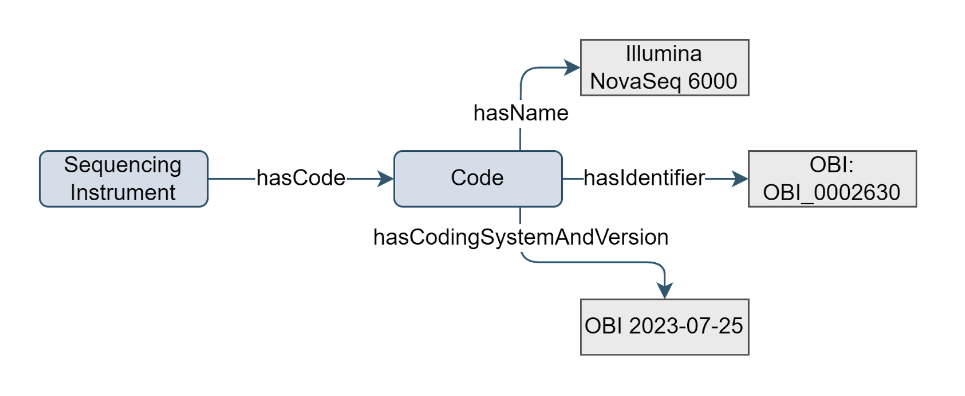
Figure 5. Example of the Sequencing Instrument concept
Guidelines for data delivery
Sequencing Instrument concept has a
hasCodeproperty which can have a value that is a descendant of:OBI:0400103 | DNA sequencer |,EFO:0003739 | sequencer |, or other
Data Processing
An essential part of scientific disciplines is processing the data produced by assays to retrieve an analysis result. Especially in data-intensive domains such as omics, data processing makes up a significant part of the experiment. Usually, individual processing and analysis steps are chained together into a (bioinformatics) pipeline. As part of data processing, data may be transformed from one format or structure to another, or may be subjected to computing to produce aggregates and other analysis results. To evaluate and reproduce these results, metadata on the data processing steps, such as the software/script that was used, is captured in the Data Processing concept.
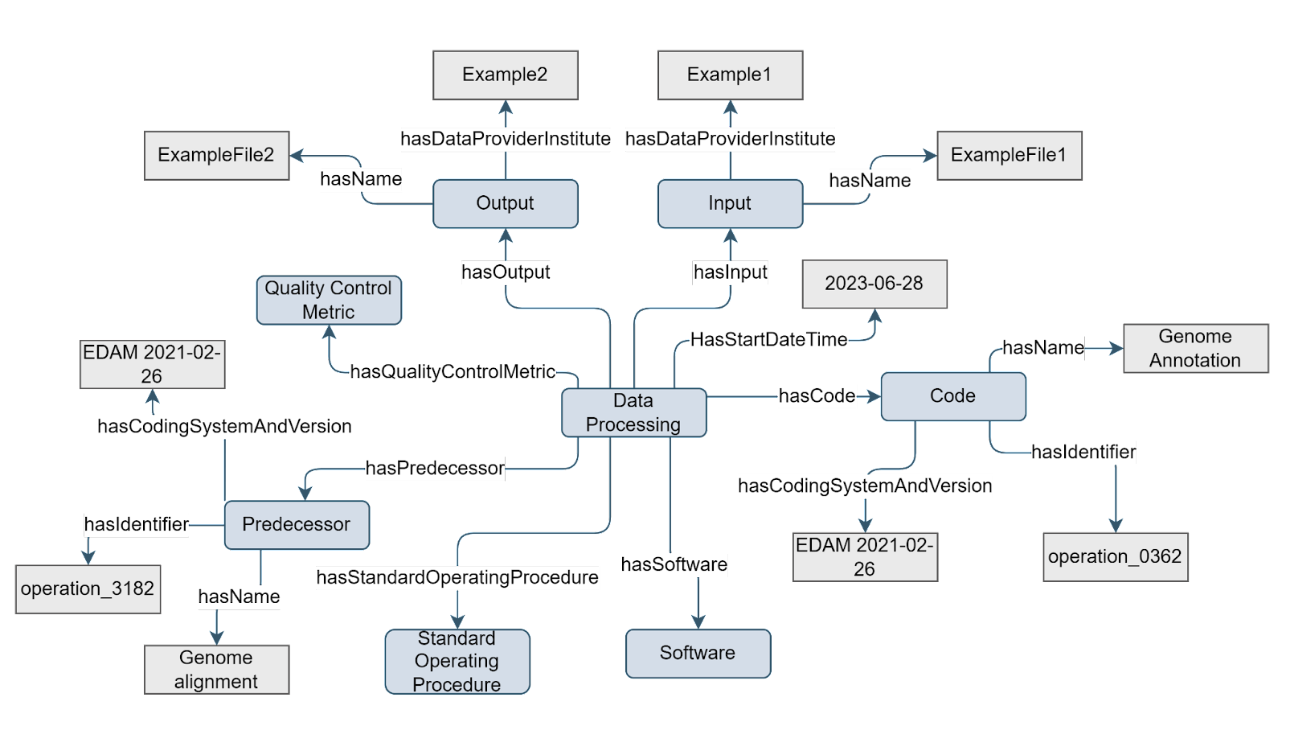
Figure 6. Example of the Data Processing concept
Guidelines for data delivery
The Data Processing concept has a
hasCodeproperty which can have a value that is a descendant ofEDAM:operation_0004 | Operation |,OBI:0200000 | data transformation |, or otherAll the properties are optional, except for the
hasCodepropertyThe Data Processing concept can be used for any data processing step for which the used software should be indicated, such as BCL to FASTQ conversion. The Data Processing concept can be used to indicate sub-steps of a broader process when there is a need to provide metadata for individual steps
Usually, a data processing step has at least one input file. However, there are cases where intermediate files between steps are not known or important. Therefore, the cardinality of
hasInputis0:n
Sample Processing
Sample processing is an essential part of the (omics) experimental workflow. It comprises all processes that manipulate a sample before it can be analysed, such as dissociating tumor cells or culturing. Some sample processing steps are characteristic for a particular omics type, such as library preparation, while others are more general, such as culturing.
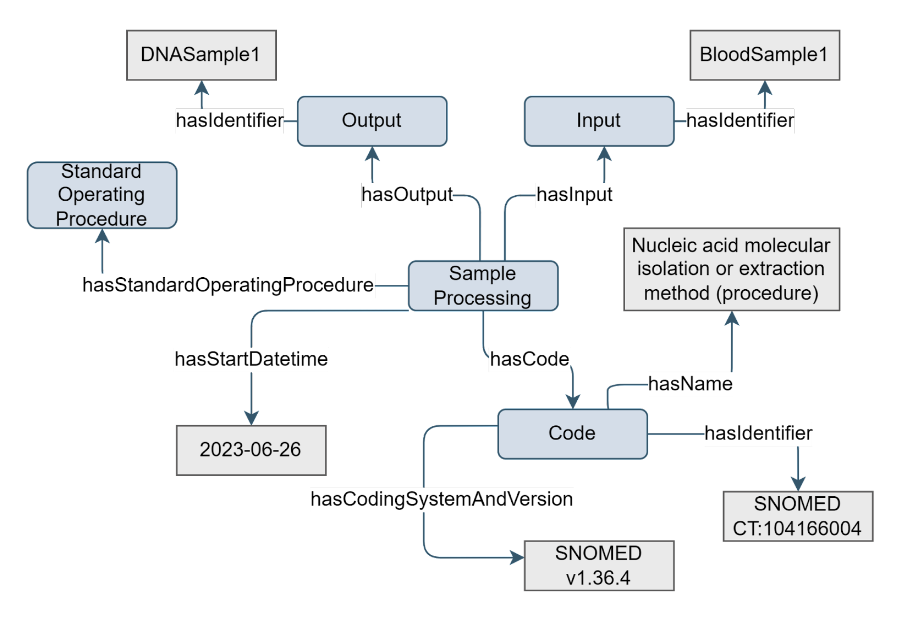
Figure 7. Example of the Sample Processing concept
Guidelines for data delivery
All properties are optional
As with all experimental processes, Sample Processing steps may be chained in sequence, and may consist of individual steps that provide additional metadata. For instance, it can be part of an Assay concept to provide essential metadata on the sample processing that is required for the particular assay type
To indicate the type of sample processing, via hasCode property, descendant terms of
EFO:0002694 | experimental process |,OBI:000011 | planned process |, andSNOMED:71388002 | Procedure (procedure) |can be usedSince Sample Processing has input and output of type Sample, having a composedOf named ‘sample’ is ambiguous. Hence the properties are named
hasInputandhasOutput. Since input and output may not always be relevant or known, for instance in case of intermediate samples between two processing steps, the minimum cardinality of these properties is 0. Note that a sample processing step may have multiple input samples, for instance in the case of a tumor sample and antibody sample, or when multiple input samples are pooled into the same library.There could also be multiple output samples for a Sample Processing. For some scenarios, it is practical to have multiple samples as output of a sample processing step. For example, in the case of creating multiple isolates from a given sample. While in the other scenarios, particularly in the context of genomics, it is prudent to to have a Sample Processing instance for each corresponding output sample.
hasStartDatetimeis an optional attribute to the Sample Processing concept. As collection datetime is mandatory for Samples, the collection datetime for the output sample is equal to the start datetime of Sample Processing
Sequencing Analysis
NGS sequencing produces raw sequencing data that should be processed and analysed. There are many options to perform this processing and analysis, such as different bioinformatics pipelines and/or scripts (commonly referred to as Software). Metadata about which pipeline and version was used, as well as the used reference genome, are important to compare and evaluate the sequencing results. The Sequencing Analysis concept, a specific type of Data Processing, can be used to store this metadata.
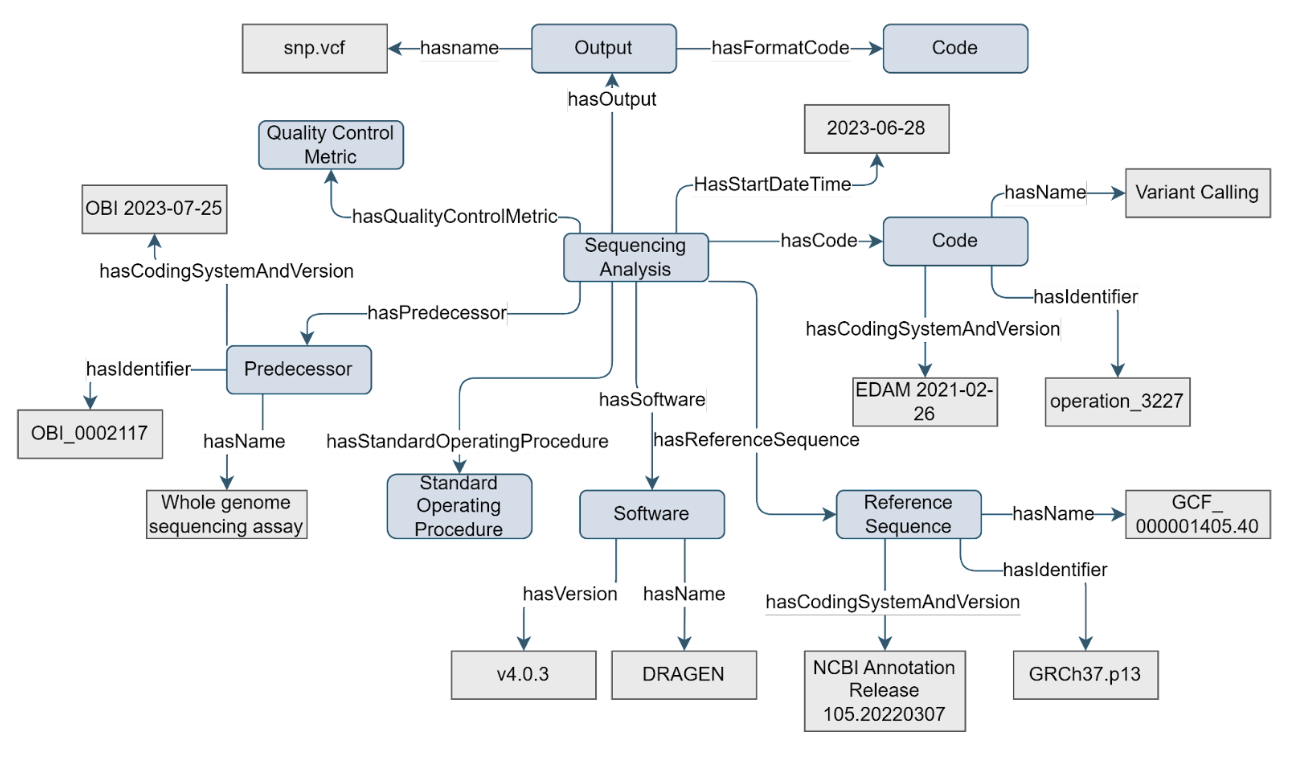
Figure 8. Example of the Sequencing Analysis concept
Guidelines for data delivery
Sequencing Analysis may have many Data Processing parts that are executed one after the other, which can be Sequencing Analysis parts themselves, such as alignment to a reference genome, or more general Data Processing parts, such as data transformation from SAM to BAM files
The Sequencing Analysis concept has a
hasCodeproperty which can have a value that is descendant ofEDAM:operation_2945 | Analysis |or otherThe
hasInput,hasOutput, andhasCodeproperties are mandatory, while everything else is optionalSequencing Analysis is introduced as a special type of Data Processing that always has the aim to analyse data produced by an upstream Sequencing Assay, and uses a reference genome (except in case of de novo assembly). Reference Sequence concept represents the reference genome in case of single organism sequencing, but can be any reference in case of metagenomics sequencing
Standard Operating Procedure
The Standard Operating Procedure (SOP) is a step-by-step description of how experimental procedures should be conducted within an organisation. An SOP is characterised by its name, the textual description, and a version. The purpose of the Standard Operating Procedure concept is to provide the textual description as agreed upon within an organisation, for the protocol followed in an experimental process.
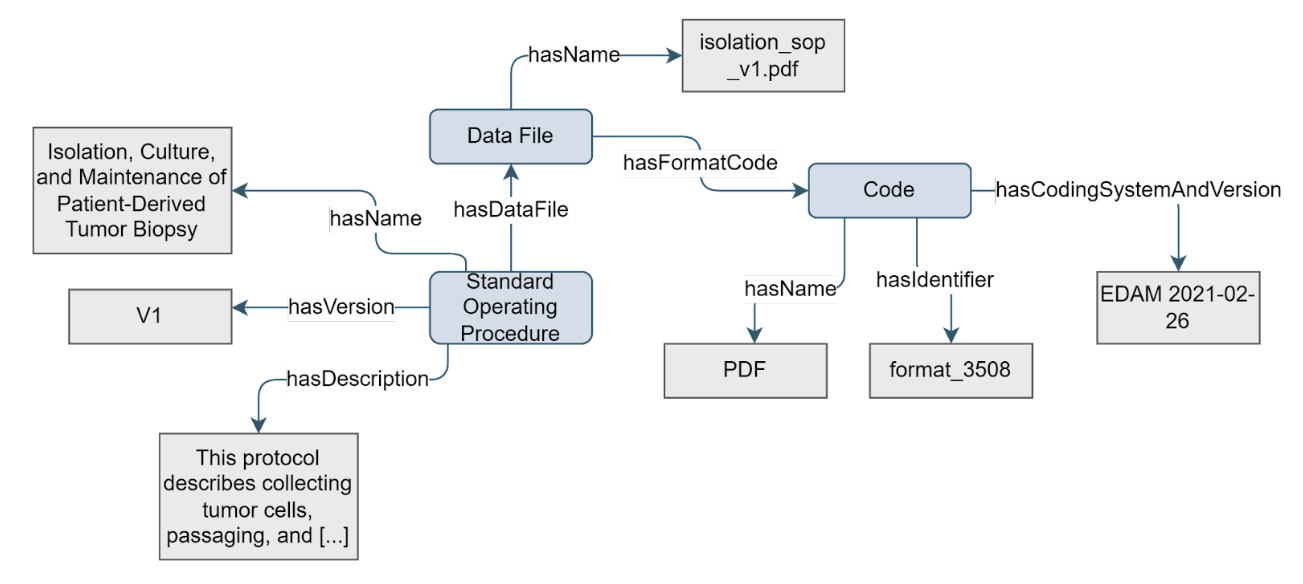
Figure 9. Example of the Standard Operating Procedure concept
Guidelines for data delivery
All properties, except
hasDataFile, have a1:1cardinality. Which means that the SOP must have exactly one name, version and descriptionThe documentation of the experimental steps to follow, or that has been followed, can be indicated as Protocol, or more broadly as Standard Operating Procedure (SOP). SOP has a notion that it is prescribed by an organisation. It also may be broader than experimental protocols
SOPs are available within organisations, but are usually available as documents or text that is not typed or classified, apart from an indication in its name. The experimental process that it prescribes and that it is linked to implicitly types the SOP
Library Preparation
The Library Preparation concept is a type of Sample Processing that is part of a Sequencing Assay. It holds information on the library preparation kit, target enrichment kit, intended insert size, and, in case a gene panel kit is used as target enrichment, information on the gene panel’s focus genes. Any other processing steps that precede an assay’s library preparation may be registered using the Sample Processing concept.
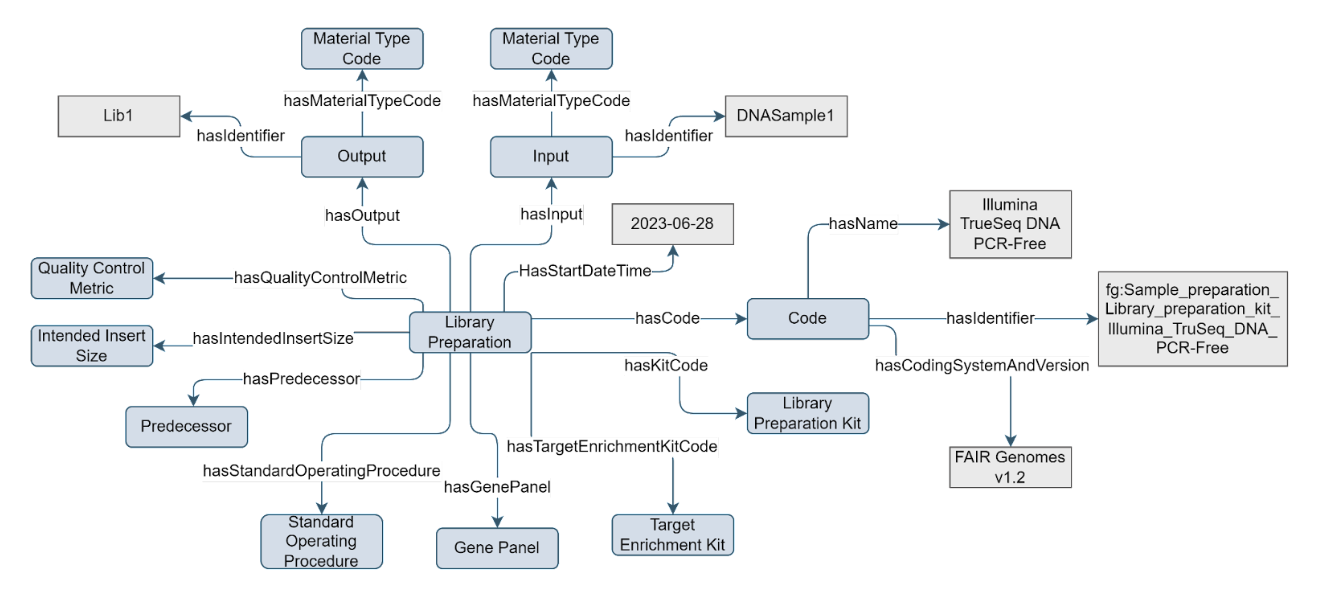
Figure 10. Example of the Library Preparation concept
Guidelines for data delivery
The Library Preparation concept has a
hasCodeproperty which can have a value that is a descendant ofOBI:0000711 | library preparation |, or otherLibrary Preparation can be considered an integral part of performing an NGS sequencing experiment. However, since the configuration/parameters of these experimental processes can vary separately, and also because they can be executed at different facilities at different times by different people, while also having their independent quality control metrics, these are represented as separate concepts that are part of the Sequencing Assay. Note that the linked Sequencing Assay and its Sequencing Instrument are tightly bound to this concept, because these influence the possible choices for the kits used for library preparation and constrains the intended read length and insert size
Note that as with other (experimental) processes, individual sub-steps can be provided, for instance for DNA extraction or amplification
All properties have a cardinality of either
0:1or0:n. This means all properties are optional.
Quality Control Metric
Quality control metrics are used to express the quality of a product or process. They help identify defects, errors, or deviations from established standards. By capturing these metrics in a Quality Control Metric concept, users can ensure that the data meets the quality criteria.
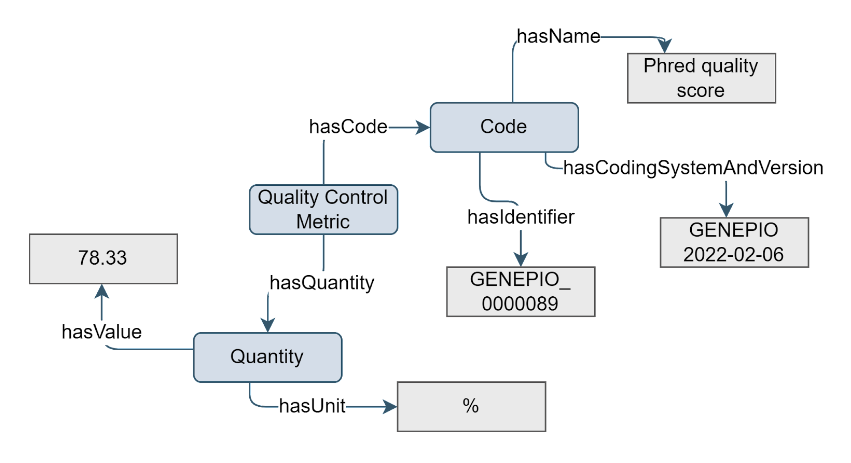
Figure 11. Example of the Quality Control Metric concept
Guidelines for data delivery
Some of the concepts, like Sequencing Run, require a Quality Control Metric. Note that although this information can be mandatory, it is not necessarily guaranteed that data quality will meet or exceed a specific standard; it still needs to be evaluated by the data user, and not all users hold on to the same data quality standards
One or multiple *Quality Control Metric*s can be assigned to a single concept (where applicable)
Isolate
The Isolate concept captures information about specific isolates and their characterization. The Isolate concept is a type of Sample concept.
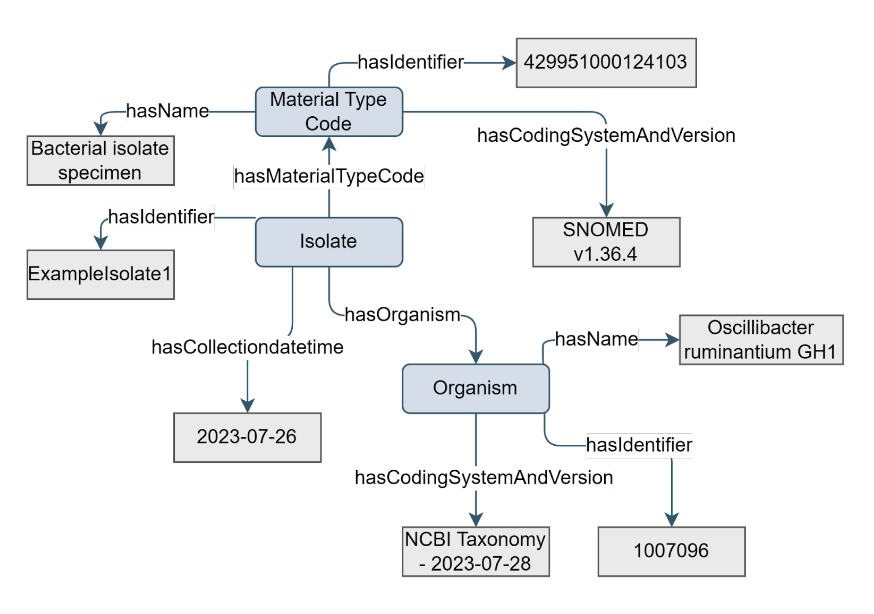
Figure 12. Example of the Isolate concept
Guidelines for data delivery
In contrast to the Sample concept, the Isolate concept is always defined by a species and strain of the pathogen/microbe that is isolated and not of the host
Gene Panel
Gene panels are used for targeted screening in both clinical and research applications. When a gene panel is used for target enrichment as part of Library Preparation, information on the gene panel and its focus genes are required to interpret downstream results. Therefore, the Gene Panel concept can be used to add metadata on the focus genes of the panel to the Library Preparation concept, which is part of a Sequencing Assay.
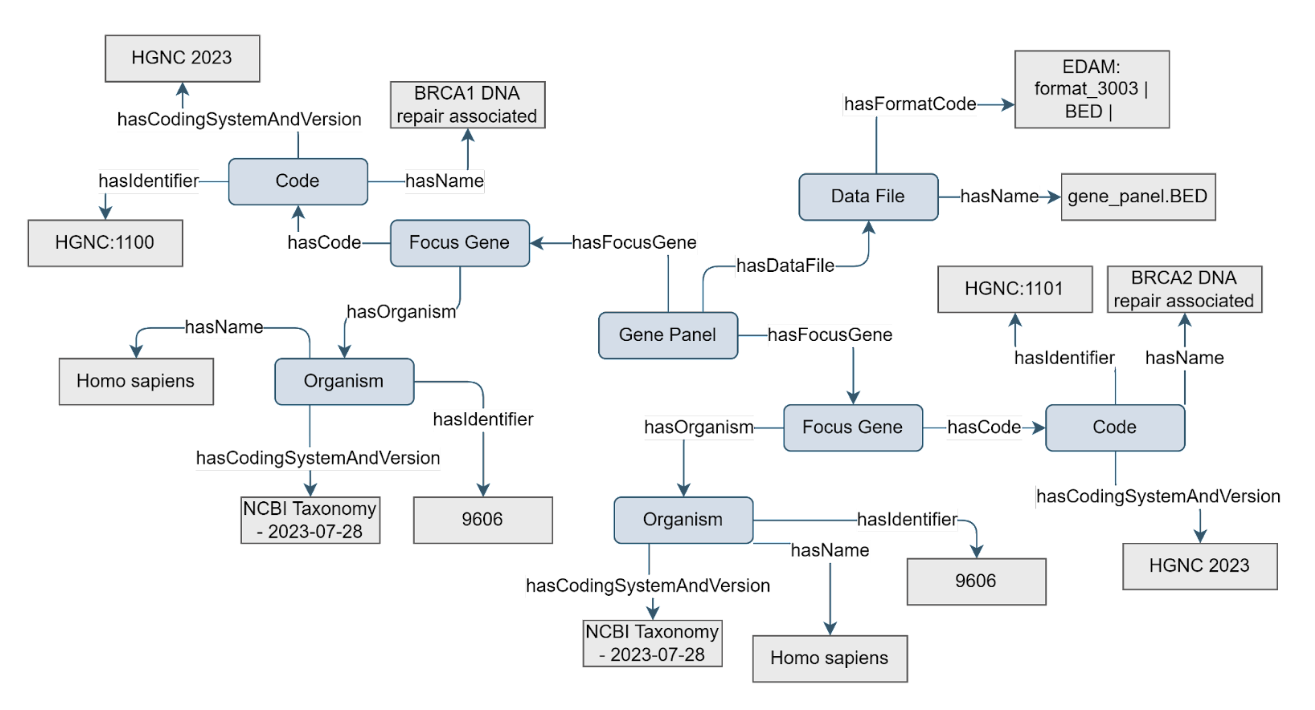
Figure 13. Example of the Gene Panel concept
Guidelines for data delivery
The Gene Panel concept must have at least 1 associated focus gene via the hasFocusGene property
Example of semantic inheritance
For different types of omics research, different types of assays will be relevant, each with their unique set of properties. Therefore, the Assay concept exists. One example of model extension is with a Mass Spectrometry Assay.
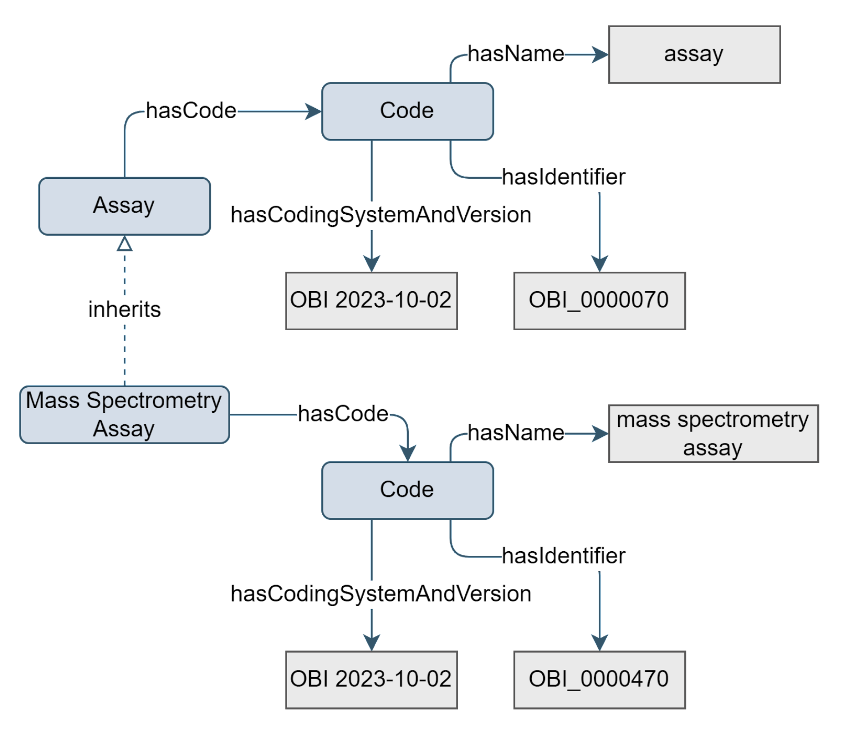
Figure 14. Example of the Assay concept inheritance
Representation of genetic variants
The set of concepts encompassing genomic variants provides the necessary foundational elements for representing variants in a concise and machine-readable format. The chosen representation draws inspiration from GA4GH Phenopackets and GA4GH VRS, aligning with the logical framework of the widely adopted HGVS variant description nomenclature.
Concept design
The design of the concepts for the representation of genomics variants follows the pattern introduced by the VRSATILE framework (a set of conventions extending GA4GH VRS) where descriptors function as central concepts providing all metadata of a specific value concept. In accordance with this design, the Variant Descriptor concept enables the description of various genomic variations, ranging from simple single point mutations to more intricate structural variations involving large genomic regions. A Variant Descriptor can be linked to a specific variant, represented by the Genomic Variation concept. The latter serves as an umbrella concept, from which all specific variant types inherits from (see Figure 1).

Figure 1. General design of the concepts representing genomic variants.
Variant Descriptor
The Variant Descriptor concept provides a high-level, human-readable way to describe a genetic variant. It not only captures essential details such as variant type, zygosity, and mutation origin, but also enables direct reference to known variants in repositories like ClinVar or RefSNP through its code attribute, thus ensuring easy linkage to well-documented variants. Genomics variants are commonly described using text strings of varying complexity and according to specific nomenclatures such as HGVS, SPDI, or ISCN in the case of structural alterations impacting chromosomes. To accommodate this diversity, the Variant Descriptor concept can be linked with one or more instances of Variant Notation, a flexible concept enabling the representation of a variant using a simple string and a reference notation.
Representing genomic variants using different notations and, consequently, different syntaxes requires users to parse such text strings to effectively query and compare data from diverse sources. For this reason, variants can be linked to the Variant Descriptor in a machine-readable manner through the Genomic Variation concept and its children concepts.
Playing the role of central concept for variant representation, the Variant Descriptor directly connects with the Source System, the Administrative Case, the Data Provider, and the Subject Pseudo-Identifier concepts (see Figure 2).

Figure 2. Design of the Variant Description concept showcasing the connection with Variant Notation and the umbrella concept Genomic Variation.
Genomic Variation
The concept of Genomic Variation serves as a generic umbrella term for a series of concepts describing genomic variations types. Genomic Variation is an empty concept and should never be directly instantiated. Instead, one of the child concepts (see figure 1) should be used. The following section will provide detailed explanations for each of the specific concepts covering the most commonly found variants types.
Single Nucleotide Variation
As the name suggests, the Single Nucleotide Variation concept covers subtle genetic alterations that occur at the level of individual nucleotides within a DNA sequence (SNVs). These variations involve the replacement of one nucleotide with another, such as adenine (A) being substituted for guanine (G) or cytosine (C) for thymine (T) at a precise location within the sequence. An example of data delivery is shown in figure 3.

Figure 3. Example of instantiation for the Single Nucleotide Variation. This example showcases the ClinVar variant VCV001684428.1. .
Genomic Insertion
The Genomic Insertion concept refers to genetic alterations characterized by the addition of one or more nucleotides at a specific location within a DNA sequence. In its current implementation, the design fully supports insertions of simple, contiguous sequences. However, more complex insertions, such as those involving inverted or duplicated sequences, as well as insertions into unknown loci, are not supported. Figure 4 illustrates an example of how this data is presented.
Note 1: The Genomic Insertion concept can describe insertions at either the sequence or chromosomal level. The property hasFeatureLocation can be specified as either a Genomic Position or a Chromosomal Location, depending on the nature of the insertion being described.
Note 2: Figure 15 shows an example of how a complex variation could be implemented. An instantiation of Genomic Insertion could indeed capture the same information albeit with a loss of metadata.

Figure 4. Example of instantiation for the Genomic Insertion concept. This example showcases the ClinVar variant VCV001684356.1.
Genomic Deletion
The Genomic Deletion concept refers to genetic alterations involving the removal of one or more nucleotides at a specific location within a DNA sequence or large deletions at chromosomal level. Currently, the design fully supports the deletion contiguous sequences and is not well suited for describing more complex cases, such as deletions spanning exon-exon, intron-exon, or exon-intron junctions, as well as mosaic or chimeric scenarios. Figure 5 provides an example of how this data is represented.

Figure 5. Example of instantiation for the Genomic Deletion Concept. This example showcases ClinVar variant VCV002445626.1.
Copy Number Variation
The Copy Number Variation (CNV) concept refers to genomic changes that alter the number of copies of a specific DNA segment within an individual’s genome. These variations can include both deletions and duplications, resulting in deviations from the normal number of copies of genomic regions. These changes manifest as differences in the amount of entire genomic segments. Examples include the deletion or duplication of entire genes or larger chromosomal regions. An example of data delivery is shown in figure 6.
Note 1: In some contexts, especially in clinical reports or preliminary research findings, the focus lies on the most clinically relevant aspect of a variant such as the presence of a CNV affecting a specific gene. This often led to simplified reports where the number of copies of a gene is highlighted, but detailed mapping of the CNV’s boundaries may be omitted. To accommodate this requirement, the cardinality of the property hasFeatureLocation has been set to 0:1. This allows to specify a list of affected genes (Variant Notation -> hasGene) alongside minimal information about the number of copies, without having to specify the precise boundaries of the CNV.

Figure 6. Example of instantiation for the Copy Number Variation Concept. This example showcases ClinVar variant VCV003063561.1.
Gene Fusion
Gene fusions can be divided in two main groups: Chimeric transcript fusions and regulatory fusions. Chimeric transcript fusions are driven by genomic rearrangements and involve two gene loci, resulting in the concatenation of segments from each into a single transcript. Regulatory fusions are rearrangements of regulatory elements from one gene near a second gene, typically resulting in the increased gene product expression of the second gene. The Gene Fusion concept capture this particularity through the property hasRegulatoryFeature. This allows to specify the type of regulatory feature (e.g. a Promoter) which is involved in the gene fusion event. Figure 7a shows an example of a chimeric fusion and figure 7b shows an example of a regulatory fusion.
In addition to that, the Gene Fusion concept use the property hasGene to capture the specific genes which are involved in the fusion event.

Figure 7. Example of the Gene Fusion concept. 7a) Example of the BCR-ABL1 fusion gene on chromosome 9. 7b) Example of a regulatory fusion with genes ERG and TMPRSS2. The promoter of TMPRSS2 is fused with the ERG gene [PMID 18165275].
Note 1: Within Variant Descriptor, the hasGene property is used to indicate genes context where the variation occurs. Given that a gene fusion creates a new genetic entity, it would be misleading to use this attribute to list the components the hybrid gene. Therefore, it is recommended to either: • Not instantiate the Variant Descriptor’s hasGene attribute in the context of Gene Fusion. • Only instantiate the Variant Descriptor’s hasGene attribute for the gene whose 5’ regulatory elements drive the expression of the fusion gene (if this information is known).
Genomic Delins
The Genomic Delins concepts covers genomic alteration where one sequence is deleted and replaced by another. A variant should be described using the genomic delins concept only if the deleted and/or inserted sequence is more than 1 bp in length. Single base pair (1 bp) substitutions fall under the Single Nucleotide Variation concept.
Unlike simple insertions or deletions, for delins mutations the exact sequences are typically provided. For this reason, the concept is only instantiated at a sequence level using Genomic Position to indicate the exact location of the variant.
An example of a Genomic Delins is shown in figure 8.

Figure 8. Example of instantiation for the Genomic Delins concept. This example showcases the ClinVar variant VCV000973581.2 and shows a deletion of 1132 bp and an insertion of 1 bp.
Genomic Inversion
The Genomic Inversion concept refers to sequence changes where, compared to the reference sequence, more than one nucleotide is replaced by the reverse complement of the original sequence. For example, AGTT → AACT would be classified as a genomic inversion. In contrast, a sequence variation such as AGTT → TCAA is not considered an inversion, but rather a genomic delins.
This concept can also be applied to represent inversions at the chromosomal level, where larger segments of DNA are reversed. Figure 9 provides an example of a genomic inversion variant.

Figure 9. Example of instantiation for the Inversion Concept. This example showcases the ClinVar variant VCV001945927.3 and shows an inversion of CA → TG.
Note 1: The property hasNucleotideSequence in the context of Genomic Inversion is used to capture the original sequence before the inversion occurred.
Note 2: Since genomic inversions can occur at both the chromosomal and sequence levels, the location of the variant should be specified accordingly—either as a Genomic Position or a Chromosomal Location.
Genomic Duplication
The Genomic Duplication concept represents a sequence change where a sequence is duplicated and the copy is inserted immediately after the original sequence at the 3’ end. If a sequence is duplicated, but inserted not directly at the 3’ end, the variation can be described using Genomic Insertion.
This concept can also be applied to represent duplications at the chromosomal level, where larger segments of DNA are duplicated and the copy is inserted adjacently to the original region.
Figure 10 provides an example of a genomic duplication variant.

Figure 10. Example of a Genomic Duplication Concept. This example showcases the ClinVar variant VCV000974714.2 and shows the duplication of a 56053 bp sequence.
Note 1: Since genomic duplications can occur at both the chromosomal and sequence levels, the location of the variant should be specified accordingly—either as a Genomic Position or a Chromosomal Location.
Note 2: If the data do not provide evidence that the extra copy of a detected sequence is directly 3’-flanking the original copy, the variation should be described as a Genomic Insertion rather than a Genomic Duplication.
Genomic Transposition
The Genomic Transposition concept refers to variations in which a typically large segment of DNA moves from one location in the genome to another. This concept captures the information by specifying the breakpoint where the segment is removed and the breakpoint where it is inserted.
A chromosome transposition, whether involving a whole segment or part of it, can often be accompanied by additional events such as insertions, deletions, and/or inversions. These accompanying changes increase the complexity of the transposition, making it challenging to model accurately. For this reason, more complex events occurring in addition to the transposition are not directly modelled, but can be captured within the description of the variant through the property hasNotation of Variant Descriptor. Figure 11 shows an example of a simple Genomic Transposition variant.

Figure 11. Example of instantiation for the Genomic Transposition concept. This example showcases a transposition from chromosome X to chromosome 4. The Variant Descriptor with a Notation is added to show the accompanied variant notation.
Note 1: Due to their complexity, instances of Genomic Transposition should always be accompanied by an instance of Variant Notation within Variant Descriptor.
Genomic Translocation
The Genomic Translocation concept describes a chromosomal alteration where, at a specific nucleotide position (the breakpoint), all nucleotides upstream originate from one chromosome, while those downstream come from a different chromosome. In the current concept model, similar to Genomic Transpositions, both simple and complex translocations are only defined by specifying their breakpoints. The complexity of the variant is therefore captured using the property hasNotation of Variant Descriptor. Figure 12 provides an example of a complex translocation, where the variant’s complexity is detailed using the Variant Notation concept. A simple translocation is modelled in the same way; the only difference lies in the simpler notation used to represent it.

Figure 12. Example of instantiation for the Genomic Translocation Concept. This example showcases a translocation between chromosome 3 and chromosome 14. The notation shows that this complex variant also contains a delins and an inversion; this information is only captured by the Variant Notation instance.
Note 1: Due to their complexity, instances of Genomic Translocation should always be accompanied by an instance of Variant Notation within Variant Descriptor.
Note 2: In order to correctly represent a translocation event, the minimum number of breakpoints to be instantiated is 2.
Nucleotide Sequence
The Nucleotide Sequence concept is used to represent a specific sequence of nucleotides within a DNA or RNA molecule. This sequence can be provided in two forms: as a literal sequence (hasLiteralSequence), such as ‘ATTG,’ which directly specifies the nucleotide order, or as a derived sequence (hasDerivedSequence), defined by its start and end positions on a reference genome. The concept also includes the property hasSequenceLength which indicates the number of nucleotides or base pairs in the sequence, providing a measure of its size. This concept is essential in genomic data modelling, as various Genomic Variation concepts—such as insertions, deletions, duplications or inversions—rely on the Nucleotide Sequence concept to accurately describe sequence changes or rearrangements at the nucleotide level.

Figure 13. Model definition of the Nucleotide Sequence concept: This concept has three attributes—a derived sequence, represented by the Code concept; a literal sequence, represented by a string; and the sequence length, measured in base pairs (bp) for double-stranded sequences or nucleotides (nt) for single-stranded sequences and represented by the Quantity concept.
Examples for data delivery
In the first example (see Figure 14), a patient underwent genomic analysis targeting genes involved in lung cancer. One of the variants observed pertains to the Epidermal Growth Factor Receptor (EGFR) gene and results in a likely benign mutation of a single nucleotide at a specific locus. To describe this variant, it is necessary to instantiate Variant Descriptor and link this instance with an instance of the Single Nucleotide Variation concept. The Variant Descriptor instance provides comprehensive details about the variant, which, in this case, is classified as a ‘substitution’ (hasTypeCode). Since the sample used for the analysis is derived from a tumor, the allele is of somatic origin (hasAlleleOrginCode). Furthermore, the variant is observed to affect both alleles at a specific locus, indicating a ‘homozygous’ zygosity (hasZygosityCode). The allele is documented and registered in public databases such as ClinVar, with an accession number provided for easy cross-referencing (hasCode).
The variant’s computable representation is created by instantiating the Single Nucleotide Variation concept. This includes specifying the exact locus of the variation relative to a reference sequence (hasGenomicPosition), and identifying the nucleotide substitution, in this case, ‘G>T’ (hasReferenceAllele and hasAlternateAllele).

Figure 14. Example of mock instantiation of a Variant Descriptor and Single Nucleotide Variation to describe a SNV discovered in a patient genetic test.
Similar information can be provided by an instance of Variant Notation directly linked to the Variant Descriptor. In the first example, this allows providing a complementary description of the SNV using the widely known HGVS notation. However, in the second example (see figure 15), Variant Notation can be used to describe Genomic variations not covered by a specific concept. For example, complex variations are not covered by any of the genomic variation concepts. The ClinVar variation VCV000224516.3 is used as an example. This complex variation contains duplications, inversions, insertions and deletions.

Figure 15. Example of mock instantiation of a Variant Descriptor to describe a complex variation. This example showcases the ClinVar variant VCV000224516.3.
Guidelines for data delivery
Variant Descriptor
The Variant descriptor concept can be used to describe:
A candidate diagnosed variant.
A variant result of a specific molecular test (e.g., sequencing or genotyping).
A candidate variant in specific molecular test (e.g., targeted variant).
When describing a variant, an instance of Variant Descriptor should always be present. This might or not be linked to a more detailed description of the variation using Genomic Variation. The code field allows a variation to be linked to external sources like the ClinGen allele registry, ClinVar, dbSNP, dbVAR, and others.
Genomic Variation
The concept of Genomic Variation serves as an overarching framework encompassing various types of Genomic variations. Under this umbrella, specific types of Genomic variations such as Single Nucleotide Variation, Genomic Insertion, Genomic Deletion, Copy Number Variation, Gene Fusion, Genomic Duplication, Genomic Inversion, Genomic Delins, Genomic Transpositions and Genomic Translocations are categorized.
Genomic Variation itself should not be directly instantiated but rather serves as a parent concept, guiding the organization and understanding of specific Genomic variations.
Proper usage entails employing the specific child concepts to describe Genomic variations.
Genes
The Gene attribute at Variant Descriptor specifies the gene context where the concept occurs. This is fundamentally different from the Gene attribute described in e.g. Copy Number Variation or Gene Fusion, where it describes the affected genes. Thus, genes described at those different levels do not necessarily have to be identical.
Genomic positions and chromosomal locations
In the current model, locations or loci are defined as precise positions within a sequence, with specific start and end numerical coordinates, or broad locations represented by chromosomal bands. This information is conveyed through the concepts of Genomic Position and Chromosomal Location, which are present in all the specific variant types:
Genomic Position should be instantiated when the exact coordinates of a variation are known.
Chromosomal Location is intended for representing cytogenetic results (e.g., karyotyping).
It is possible to omit positional information altogether when it is unknown.
Chromosomal Location
In the current implementation, Chromosomal Location is intended solely for human chromosomes. For use with different species, both the nomenclature for cytoband representation (currently ISCN) and the standard for describing chromosomes (currently SNOMED CT) should be extended.
For events occurring within a specific cytoband, both the start cytoband and end cytoband must be instantiated with the same value.
For events spanning across multiple chromosomal locations (e.g., large deletions), the interval must represent a contiguous region within the same chromosome. According to GA4GH VRS, the order in which cytoband coordinates are represented is p-terminus → centromere → q-terminus orientation. Consequently, bands on the p-arm are represented in descending numerical order when selecting cytobands for start and end.
Genomic Position
The concept of genomic position allows for the precise representation of coordinates within a continuous reference sequence defined by the Reference Sequence concept. An essential feature of this concept is the ability to choose the preferred coordinate system by instantiating the “Coordinate Convention” attribute, which can be set to either “Residue” or “Inter-residue” coordinate conventions (see Figure 16):
Residue Coordinate Convention: When the Residue coordinate convention is selected, each nucleotide is assigned a specific position along the sequence. This convention, commonly used in systems like HGVS or VCF, provides a straightforward representation of nucleotide positions.
Inter-residue Coordinate Convention: In contrast, the Inter-residue coordinate convention, introduced with the GA4GH VRS, defines positions between nucleotides. This system is particularly useful in scenarios involving deletions or insertions, as it offers a more precise representation of genomic coordinates and reduces ambiguity in genomic data interpretation.
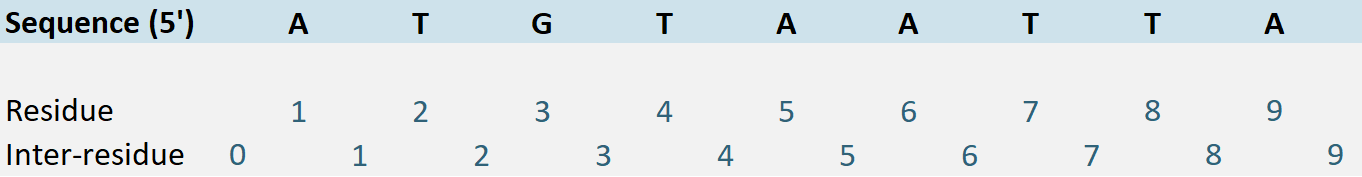
Figure 16. Coordinate conventions in use within Genetic Position.
In addition to that, the following rules applies to concept:
The minimum range for both Start and End attribute is ‘0’.
The End attribute value must be greater o equal to the Start attribute.
As for GA4GH VRS, Genomic Position consider that all locations are with respect to the positive/forward/Watson strand.